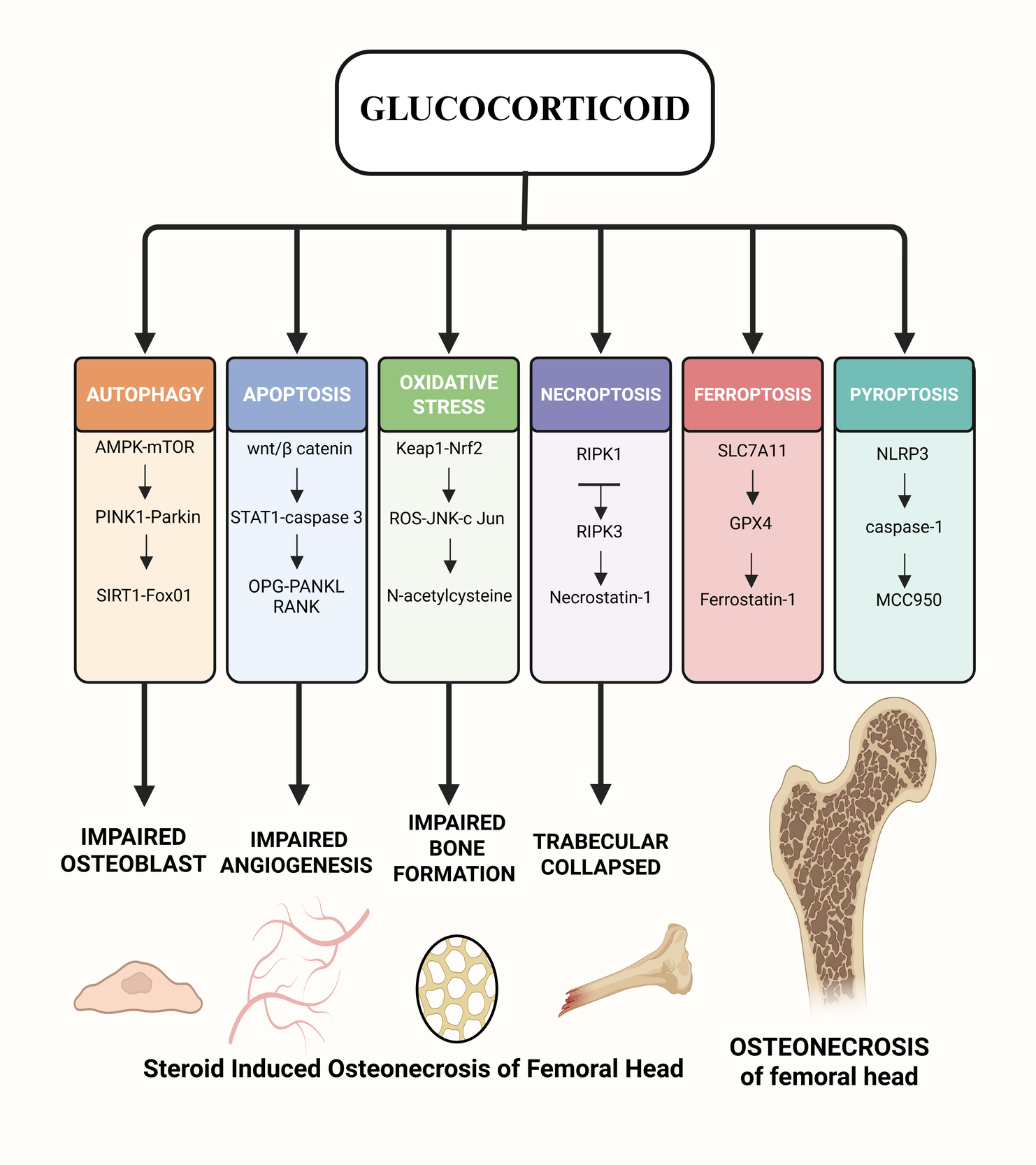
Graphical Abstract
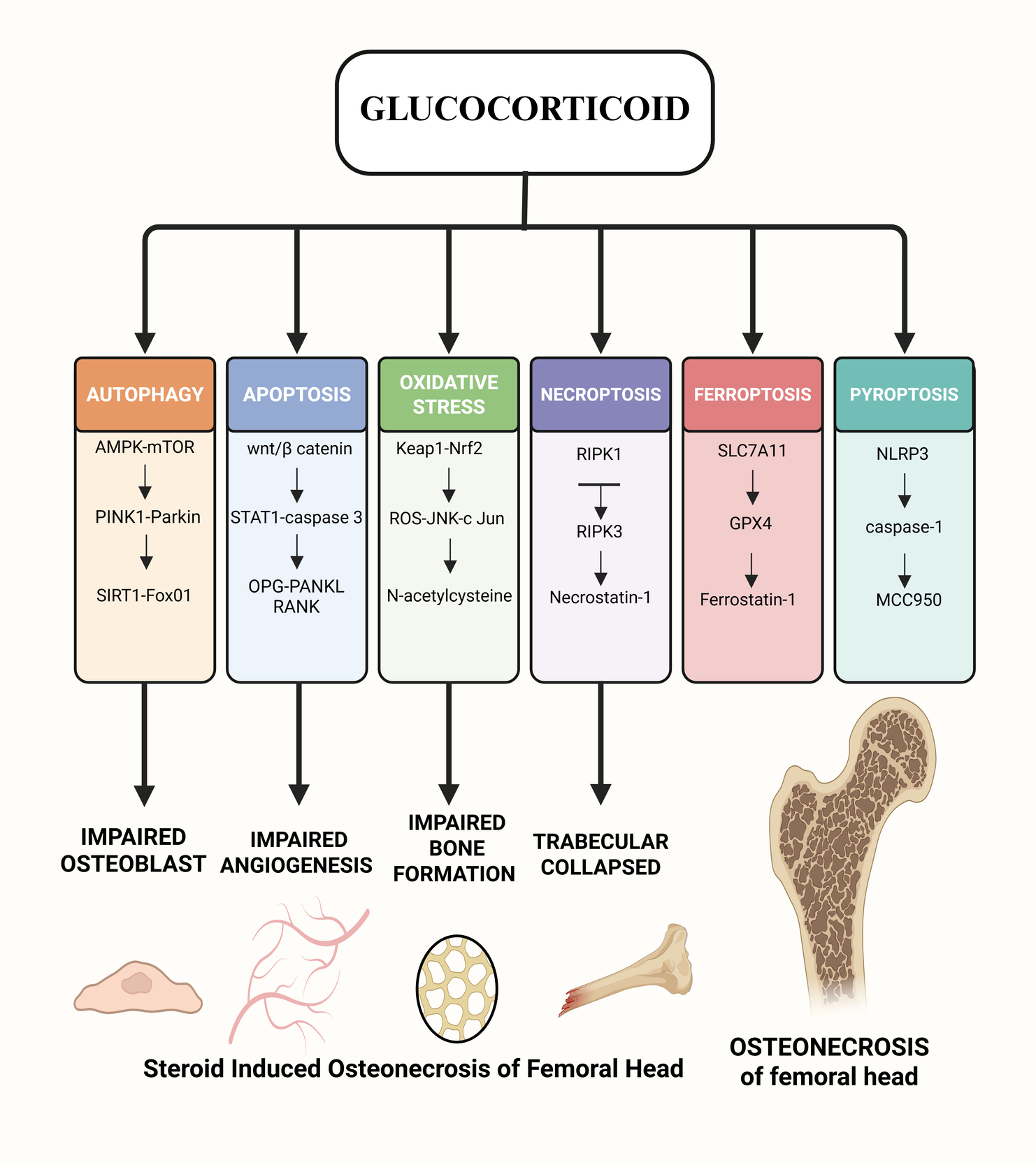
1. Introduction
Osteonecrosis of the femoral head (ONFH) is a common orthopaedic condition that is hard to cure. It has many causes, but the major symptoms are hip pain and problems with movement.1 People between the ages of 41 and 60 are more likely to get it, with a male-to-female ratio of 2.4:1. Men are diagnosed earlier than women.2 In China, there are about 8.12 million people aged 15 and up who have non-traumatic ONFH.3 SONFH, or steroid-induced osteonecrosis of the femoral head, is the most frequent kind of non-traumatic ONFH. It is a metabolic condition induced by long-term or short-term use of glucocorticoids (GCs), with femoral head endothelial cell injury, lipid metabolism abnormalities, coagulation malfunction, and cell necrosis and apoptosis being the predominant clinical signs.4 Studies have revealed that if people take 0.04g of GCs every day for more than three months, 5% to 40% of them will get SONFH. For every 0.01g increase in GCs dose, the number of people with SONFH will go up by 3.6%. The frequent occurrence of new coronavirus pneumonia in recent years has led to the widespread use of GCs. This has caused the incidence of SONFH to rise year after year, and the age of onset to tend to be younger.5,6 SONFH’s pathophysiology is rather complicated and happens because of the combined action of several signalling pathways. This article looks at the progress of research on several molecular signalling pathways that may have a role in the development of SONFH.
2. Method
2.1. Strategy for Searching the Literature
This review was performed as a thorough narrative examination of the literature regarding molecular signalling pathways in the etiology of steroid-induced osteonecrosis of the femoral head (SONFH). We conducted a thorough literature search across three principal databases PubMed, Scopus, and Web of Science, to find all pertinent papers. The search approach incorporated keywords and medical subject headings, including “steroid-induced osteonecrosis of the femoral head,” “pathogenesis,” “signalling pathway,” “apoptosis,” and “osteogenesis.” These phrases were employed in diverse combinations to guarantee thorough coverage of studies concentrating on the molecular mechanisms driving SONFH. We did not set any limits on the publication date or study design at first because we wanted to include both old and new studies on SONFH pathways.
2.2. Requirements for Eligibility
When we made the inclusion and exclusion criteria, we focused on picking research that were most relevant to SONFH molecular mechanisms:
- Inclusion criteria: We considered original research articles (experimental or clinical investigations) and review papers that particularly explain or address molecular signaling pathways related in the development or progression of SONFH.
- Exclusion criteria: We excluded publications not in English, studies not directly associated with SONFH molecular pathways (e.g., papers on osteonecrosis lacking references to steroid-related mechanisms or molecular pathway analysis), and any duplicate or overlapping publications identified during the search.
2.3. Choosing Studies
After the database searches were done, all of the records that were found were put into a reference management tool, and any duplicates were deleted. We then used a two-step screening approach to choose the final list of studies. In the initial phase, we evaluated the titles and abstracts of all distinct papers to eliminate those evidently unrelated to SONFH pathophysiology or molecular pathways. In the second stage, we got the full texts of all the other articles and checked them to make sure they satisfied the inclusion criteria and didn’t satisfy any of the exclusion criteria. We found a total of 12 papers that met all of our criteria through this systematic selection approach. These 12 study were included in the qualitative synthesis of our review.
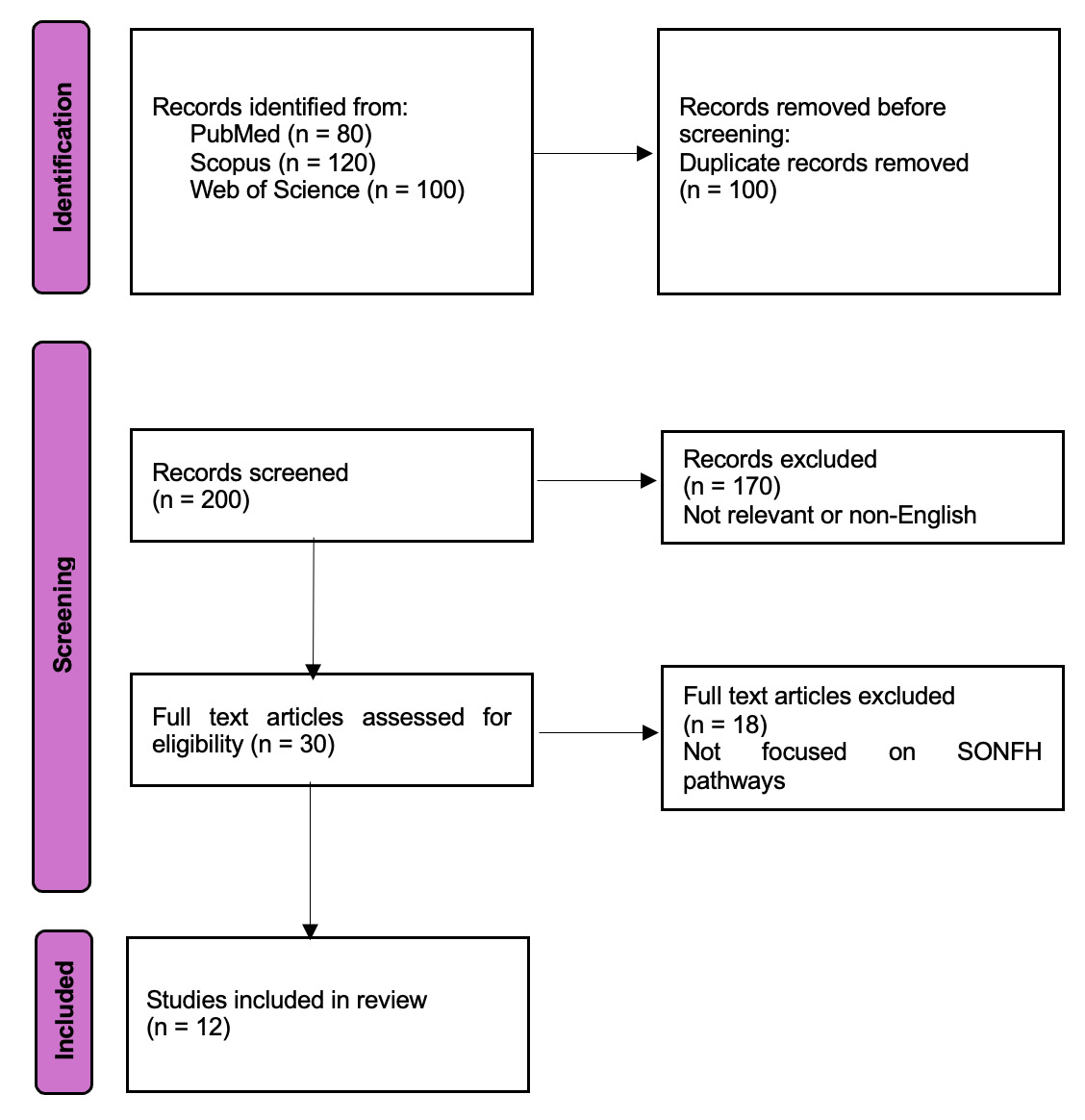
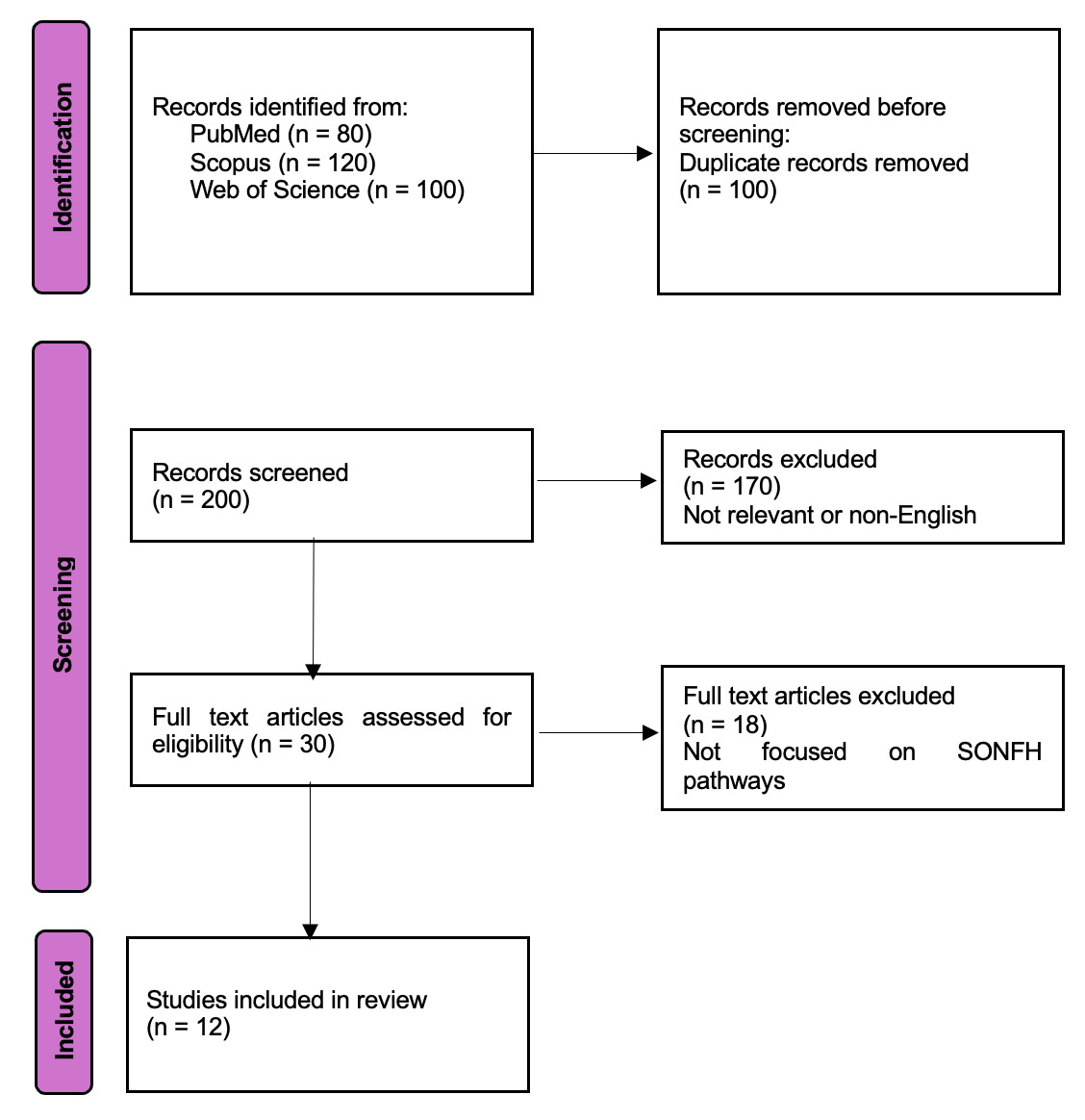
We have used a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram (Figure 1) to show how we found and chose the studies. The flow chart shows how many records were found through database searches, how many were screened at each stage, how many were not included (with short reasons for exclusion, like “irrelevant topic” or “non-English”) at the title/abstract and full-text screening stages, and how many studies (n = 12) were finally included in the review. This clear record of the search and selection procedure makes it possible to reproduce the results and meets the demand for clarity in how we narrowed down the literature to the studies that were most relevant to SONFH molecular signalling pathways.
3. Autophagy
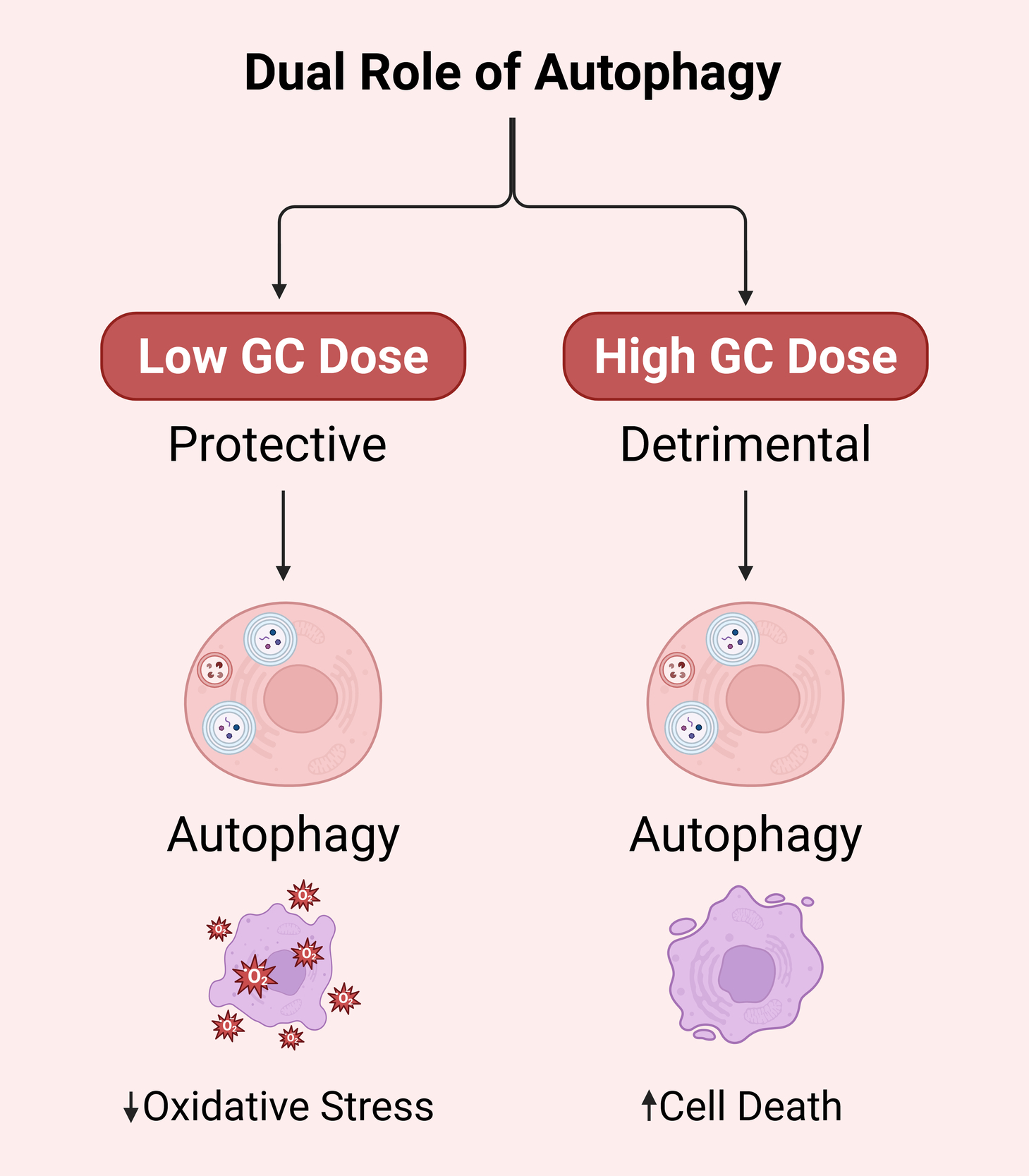
Autophagy is when cells make autophagosomes, which are microscopic structures that eat up cells that are dying or damaged and break them down into small molecules so that they can start using energy again. Cells do this to protect themselves: they keep their balance, stop apoptosis, and keep regular cell activity going even when things are tough, as when they are stressed, short on oxygen, or starving. But if the autophagy system is broken or too much autophagy happens, normal cells can die through necrosis and apoptosis.7 There have been reports that GCs can mess up autophagy, which can kill bone cells and stop them from working, which can cause and make SONFH worse.8 Researchers have recently discovered that AMPK/mTOR, PINK1/Parkin, SIRT1/FoxO1, PI3K/Akt/mTOR, and other signalling pathways are involved in SONFH.
3.1. AMPK/mTOR Signaling Pathway
The AMPK/mammalian target of rapamycin (mTOR) signalling pathway is a key approach to control cellular autophagy. AMPK keeps track of how much energy cells have. When a cell is under stress, hypoxia, or other conditions that modify the AMP/ATP ratio, AMPK is turned on and phosphorylated. This stops mTOR from working, which means that mTOR cannot phosphorylate Unc-51-like kinase 1 (ULK1). Unphosphorylated ULK1 is highly expressed, which activates Beclin-1 and LC3Ⅰ/LC3Ⅱ in significant amounts, which makes cellular autophagy happen more often.9,10 Lu et al.11 found that long non-coding RNA-TUG1 increases cellular autophagy through the AMPK/mTOR signalling pathway. This helps bone marrow mesenchymal stem cells turn into bone cells and heal bone injury. Liao et al.12 showed that activating the AMPK/mTOR signalling pathway increased the levels of autophagy-related proteins like Beclin-1 and LC3Ⅰ/LC3Ⅱ. This increased the autophagy function of femoral head endothelial cells, which helped bones grow and helped stop SONFH.
3.2. PINK1/Parkin Signaling Pathway
The homologous phosphatase tensin-induced kinase 1 (PINK1)/Parkin signalling system is a well-known signalling route that controls mitochondrial autophagy. PINK1 builds up outside the mitochondrial membrane under normal conditions. The outer mitochondrial membrane translocase and the inner mitochondrial membrane translocase then move it to the inner mitochondrial membrane, where it is broken down. High dosages of GCs can hurt mitochondria, which makes it impossible for PINK1 to get into the inner mitochondrial membrane. However, it builds up in high amounts at the outer mitochondrial membrane and activates Parkin, which makes LC3II and forms autophagosomes.13,14 Studies have shown that activating the PINK1/Parkin signalling pathway gets rid of severely damaged mitochondria in bone cells. This protects bone cells from the harmful effects of damaged mitochondria spreading throughout the body and boosts the ability of bone marrow mesenchymal stem cells to turn into bone cells.15,16
3.3. SIRT1-FoxO1 Signaling Pathway
The Silent Information Regulator 1 (SIRT1)/Forkhead Transcription Factor Subfamily O (FoxOs) signalling pathway assists in regulating cellular autophagy. SIRT1 is a deacetylase that helps control metabolism, oxidative stress, cellular autophagy, and apoptosis.17 FoxO1 is a regulatory autophagy factor in the FOX family, and SIRT1, which is higher up in the chain, controls it in a negative way. When cells are stressed, SIRT1 levels drop a lot. SIRT1 separates from FoxO1 and removes its acetylation, which speeds up the breakdown of FoxO1.18 FoxO1 can be deacetylated, which can cause the expression of autophagy-related proteins ATG7, ATG12, and Beclin-1 to go up, which starts cellular autophagy.19 Zhou Fan et al.20 observed that puerarin can help mouse osteoblasts do autophagy through the SIRT1/FoxO1 signalling pathway. This stops dexamethasone from killing osteoblasts and protects them.
3.4. PI3K/Akt/mTOR Signaling Pathway
The only signalling mechanism that has been shown to stop autophagy is the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway. This signalling pathway starts with PI3K. Cell surface receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) turn phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 fully activates Akt, which then works on its downstream target protein mTOR. This stops the expression of microtubule-associated protein 1A/1B-light chain 3Ⅰ/Ⅱ (LC3Ⅰ/LC3Ⅱ) and autophagy-related gene 1 (Beclin-1), which stops cellular autophagy.21
Jang et al.22 did cell experiments and found that methylprednisolone activated the PI3K/Akt/mTOR signalling pathway, lowered the levels of Beclin-1 and LC3 in vascular endothelial cells in the femoral head, stopped autophagy from protecting SONFH, and made SONFH worse. Wang XY et al.23 showed that blocking the PI3K/Akt/mTOR signalling pathway and starting autophagy helped stop SONFH from killing long bone cells, which is a technique to cure SONFH. But Wang Q et al.24 found that lithium turned on the PI3K/Akt/mTOR signalling pathway, lowered the levels of Beclin-1 and LC3Ⅰ/LC3Ⅱ proteins that are involved in autophagy in bone tissue, stopped osteoblasts from autophagizing too much, and helped trabeculae form, which helped stop SONFH. Guo Xuefeng and others25 did tests in rats to show that activating factors related to the PI3K/Akt/mTOR signalling pathway lowered the amount of autophagy in bone cells, which protected bone tissue and slowed the progression of SONFH. It is still not clear how to change the course of SONFH by interfering with cellular autophagy through the PI3K/Akt/mTOR signalling pathway. Also, the effects of moderately inhibiting autophagy are not the same as those of severely inhibiting autophagy.
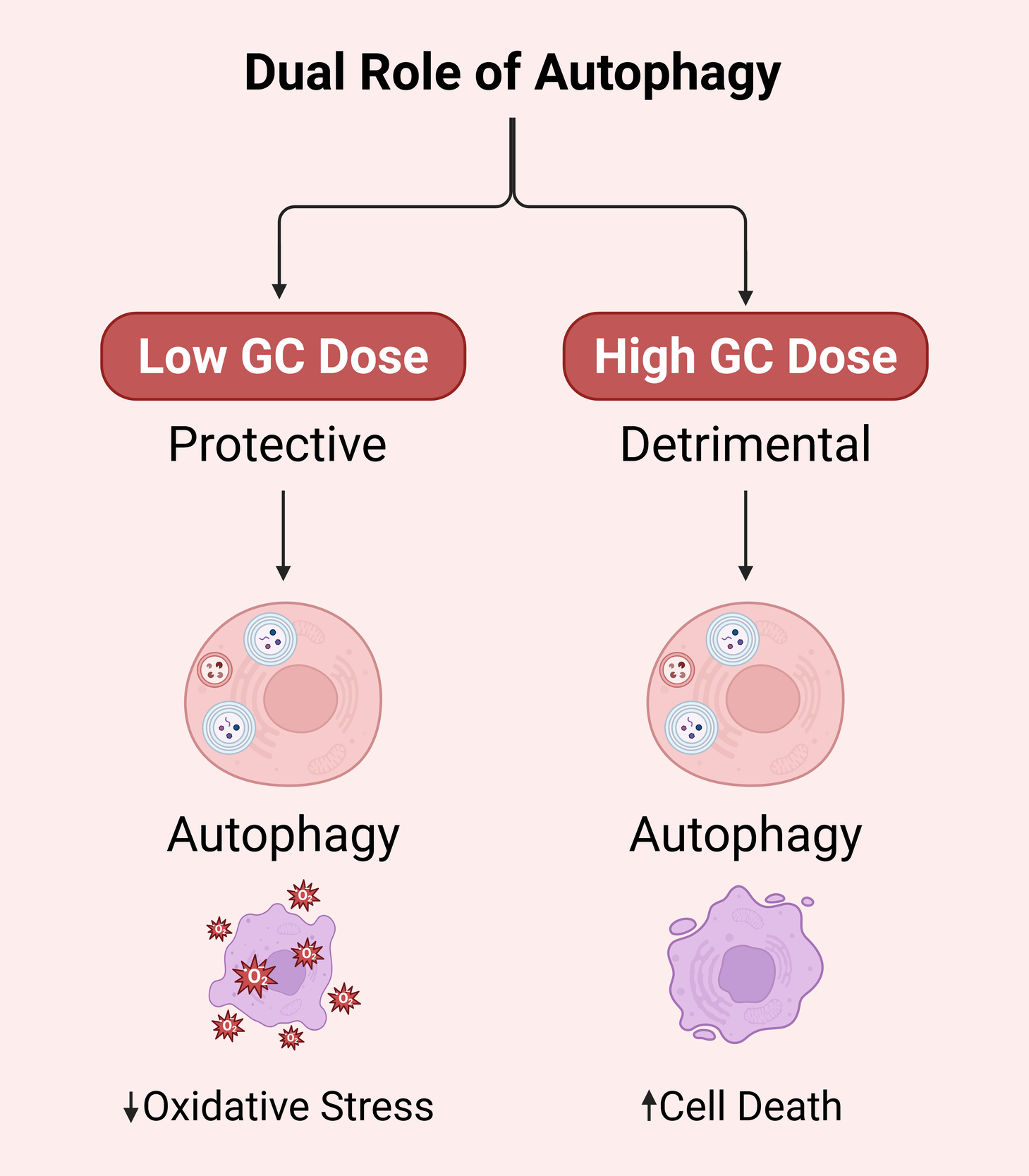
This could have something to do with the length and number of GCs. Yao et al.26 did tests on mice and showed that during the beginning of GCs treatment or at low levels (<2.8 mg/kg/d), autophagy was turned on to protect bone cells. Long-term usage of GCs or larger dosages of GCs (5.6 mg/kg/d) caused too much autophagy to build up, which led to and made worse necrosis and apoptosis in bone cells (Ilustrated in Figure 1). Lithium has been demonstrated to change the PI3K/Akt/mTOR signaling pathway, lower too much autophagy, and encourage the growth of trabecular bone. This helps stop glucocorticoid-induced osteonecrosis of the femoral head.24,26 More research is needed to find out if the smart usage of GCs and the intervention of signalling pathways, including AMPK/mTOR, PINK1/Parkin, SIRT1/FoxO1, and PI3K/Akt/mTOR, will help treat SONFH. All of these results show that autophagy plays several different roles in SONFH, and that these roles are controlled by different signaling pathways. Table 1 shows a side-by-side summary of various pathways.
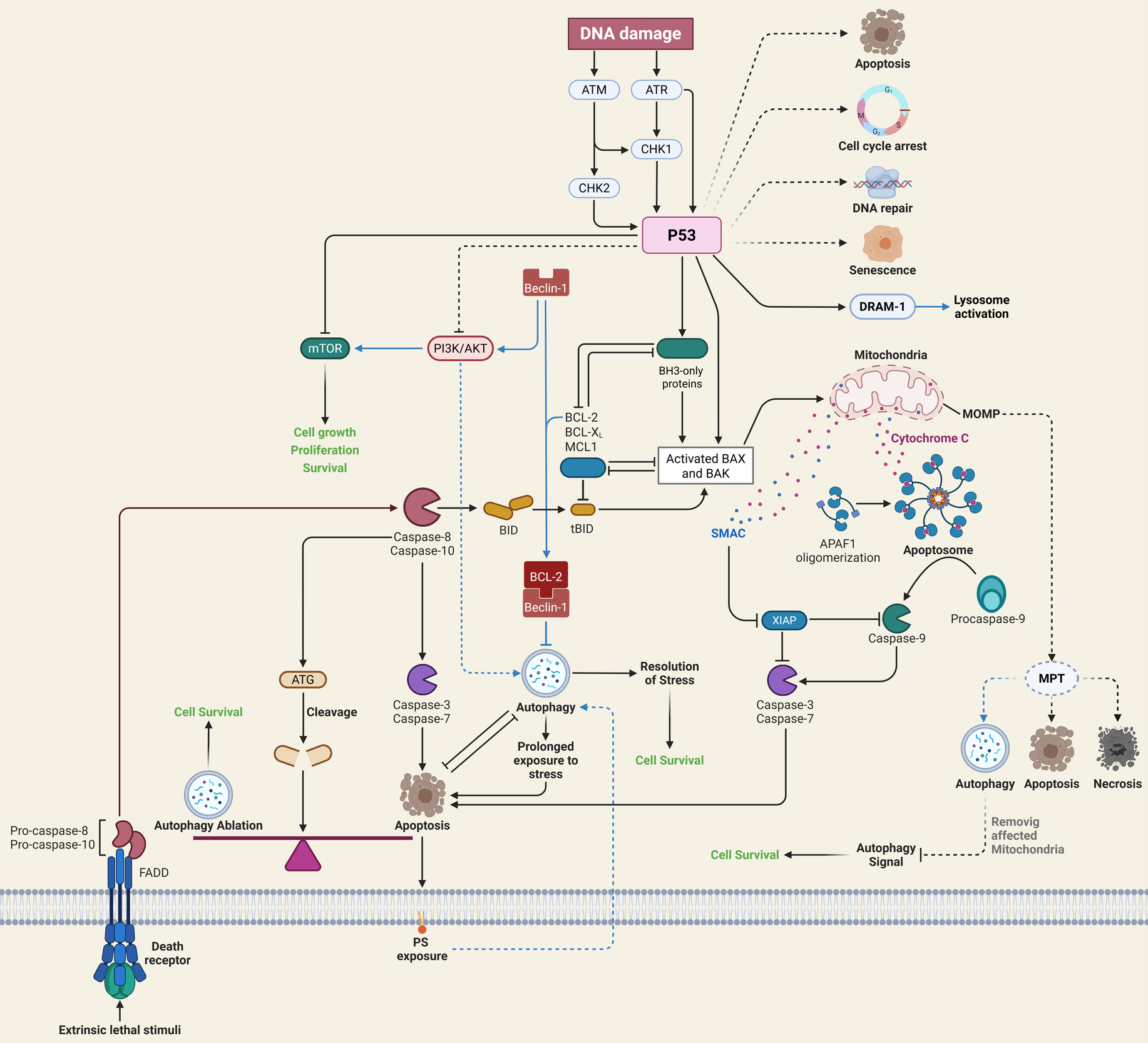
4 Apoptosis
Apoptosis is an orderly and highly regulated process of programmed cell self-destruction. It can activate the suicide program encoded by genes under the stimulation of the internal and external environment of the body, eliminate harmful cells in the body, and has great significance in the growth and development of the human body.27 Overexpression or inhibition of apoptosis will have adverse effects on the body. Apoptosis can be divided into the intrinsic mitochondrial pathway, the extrinsic pathway, and the endoplasmic reticulum pathway. All three ultimately induce cell death by activating cysteine proteases (caspases) and hydrolyzing cascade reactions.28 Studies have found that long-term use of GCs can promote osteoblast apoptosis and inhibit osteoblast formation, aggravating the induction of SONFH.29 Currently, the apoptosis pathways related to SONFH have been found to include Wnt/β-catenin, STAT1/caspase-3, and OPG/RANKL/RANK signaling pathways.
4.1. Wnt/β-catenin Signaling Pathway
The Wnt/β-catenin signaling pathway is a classic pathway for regulating cell apoptosis. It plays an important role in reducing GCs’ damage to osteoblasts, inducing osteoblast formation, inhibiting osteoblast apoptosis, and maintaining the balance of bone metabolism.30 Under physiological conditions, Wnt binds to the membrane frizzled receptor (Frz) and low-density lipoprotein receptor-related protein (LRP5, LRP6), inhibiting the degradation of β-catenin, causing β-catenin to accumulate in the cytoplasm and transfer to the nucleus, where it binds to the transcription factors T cell factor (TCF) and lymphoid enhancer factor (LEF), activating the downstream target genes of the Wnt/β-catenin pathway, such as Bc1-2, c-Myc, and cyclin D1, thereby inhibiting osteoblast apoptosis and promoting osteoblast proliferation and differentiation.31 Ren et al.32 found through rat experiments that the dried rhizome extract of Rehmannia glutinosa can activate the Wnt/β-catenin signaling pathway, inhibit osteoblast apoptosis, and play a role in preventing SONFH. Xu et al.33 found that Jintiange capsule can significantly reduce the apoptosis of osteocytes, reduce the number of osteoclasts, promote the osteogenic differentiation of bone marrow mesenchymal stem cells and the proliferation of osteocytes through the Wnt/β-catenin signaling pathway, thereby improving SONFH.
4.2. STAT1/caspase-3 Signaling Pathway
Signal transducer and activator 1 (STAT1) is an important cytoplasmic transcription factor that initiates apoptosis. Caspase-3, a cysteine-containing aspartate proteinase, is a downstream target of STAT1 and is often referred to as a death protein that regulates cell life activities.34 GCs can induce and aggravate the activation of STAT1 and phosphorylate it. Activated STAT1 leads to the dissociation of the Bcl-2-BAX complex, thereby decreasing the level of the anti-apoptotic gene Bcl-2 and increasing the level of the pro-apoptotic gene BAX, which in turn increases mitochondrial membrane permeability, leading to the massive release of cytochrome C and its combination with apoptotic protease activating factor-1 (Apaf-1), thereby activating aspartate proteinase 9 (Caspase-9). Caspase-9 can activate caspase-3, and finally lead to osteoblast apoptosis.35 Cai et al.36 found that, in vitro, erythropoietin inhibited the expression of STAT1/caspase-3 signaling pathway, reduced the expression of caspase-3 and other proteins, and promoted the expression of Bcl-2, thereby inhibiting dexamethasone-induced osteocyte apoptosis. In vivo, it can reduce the formation of empty bone pits in SONFH rats, providing a new treatment for SONFH.
4.3. OPG/RANKL/RANK Signaling Pathway
The osteoprotegerin (OPG)/receptor activator of nuclear factor κB ligand (RANKL)/receptor activator of nuclear factor κB (RANK) signaling pathway is an important pathway for regulating osteoclast proliferation and apoptosis. RANKL is a type II transmembrane protein that mediates bone resorption. RANK is a type I transmembrane protein and the only receptor for RANKL. On the one hand, RANKL activates NF-κB after binding to RANK, causing high expression of anti-apoptotic genes such as Bcl-2 and Bcl-xl, thereby inhibiting osteoclast apoptosis. On the other hand, RANKL can directly act on osteoclast precursors after binding to RANK, promoting the differentiation and maturation of osteoclasts.37 OPG is a soluble secretory glycoprotein that can inhibit osteoclast proliferation and activation. Its structure is similar to RANK and can competitively bind to RANKL, reducing the binding of RANK to RANKL, thereby inhibiting osteoclast formation, reducing bone resorption, and promoting bone remodeling.38 Yu et al.39 found that cucumber seed peptide inhibited RANKL-induced osteoclast formation and improved bone structure loss in rats through the OPG/RANKL/RANK signaling pathway. Song Hongmei et al.40 found that the warming yang and tonifying kidney formula can effectively prevent and delay SONFH by upregulating the expression of OPG through the OPG/RANKL/RANK signaling pathway and inhibiting the binding of RANKL and RANK.
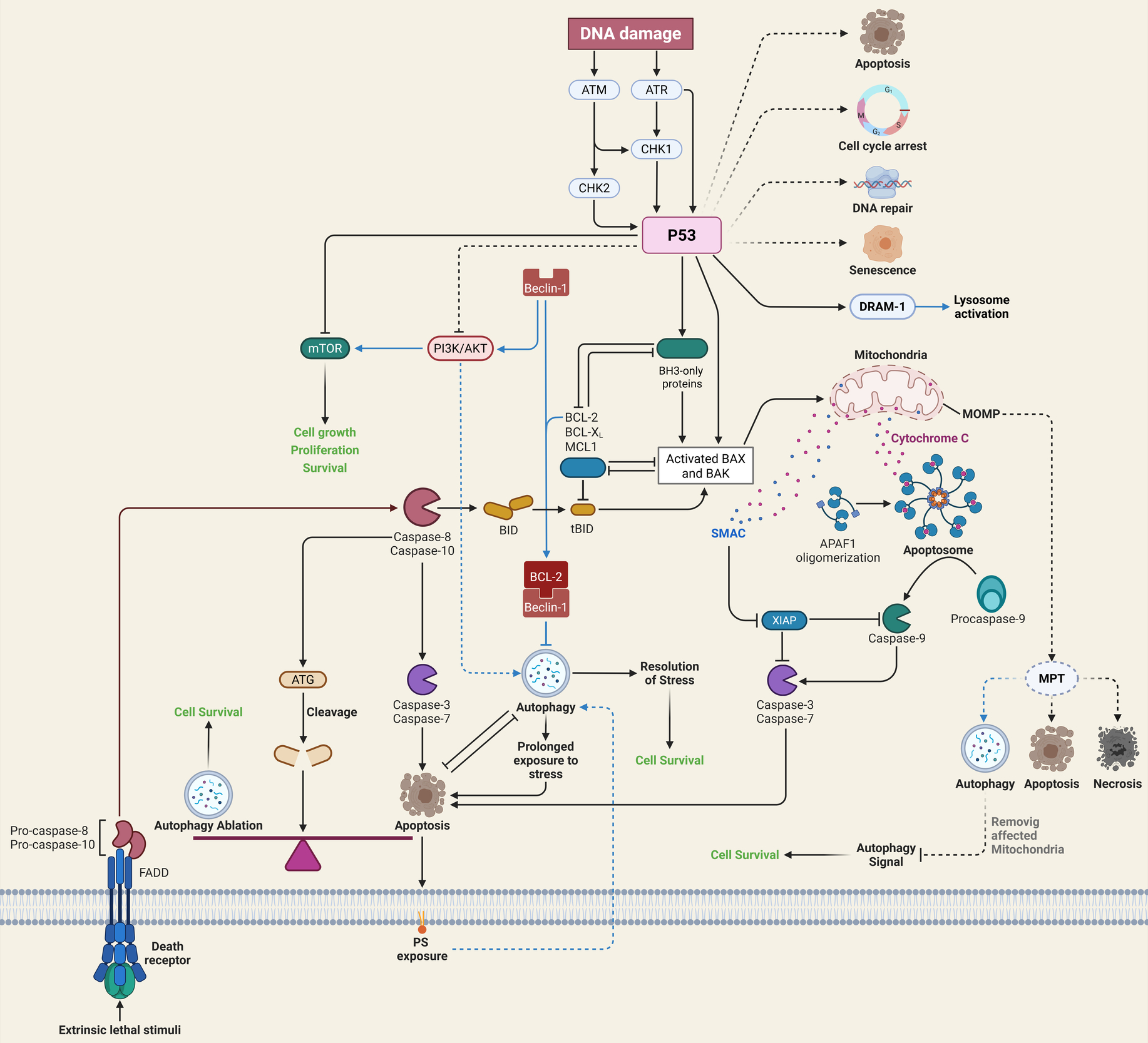
Cell apoptosis plays an important role in the pathogenesis of SONFH. Studies have found that inhibiting excessive apoptosis of bone cells through Wnt/β-catenin, STAT1/caspase-3 and OPG/RANKL/RANK signaling pathways can prevent and treat SONFH. The occurrence of SONFH may be the result of the joint participation of multiple apoptosis signaling pathways. At present, there is little research on the interaction between apoptosis signaling pathways, which needs further discussion. Wnt/β-catenin, STAT1/caspase-3, and OPG/RANKL/RANK signaling work together to keep osteoblasts alive and remodel bones in SONFH Table 2. A detailed cross talk between autophagy and apoptosis is illustrated in Figure 2 below.41
5. Ferroptosis
Ferroptosis refers to an iron-dependent programmed cell death caused by decreased cellular antioxidant capacity and massive accumulation of reactive oxygen species due to iron overload or inactivation of glutathione peroxidase 4 (GPX4).42 Different from other cell death processes such as autophagy, apoptosis, and pyroptosis, ferroptosis is iron-dependent and occurs in mitochondria, often leading to mitochondrial shrinkage and increased mitochondrial membrane density. At the same time, the cell nucleus may be of normal size, but chromatin condensation may also occur. Ferroptosis is involved in multiple metabolic pathways and is of great significance for the prevention and treatment of neurodegenerative diseases, ischemic diseases, and tumors.43 It has been proven that the ferroptosis pathway associated with SONFH is the SLC7A11/GPX4 signaling pathway.
Solute carrier family 7 member (SLC7A11) is a transmembrane protein that participates in the formation of the cystine/glutamate antiporter, maintains the balance of cystine and glutamate in the cell, and ensures that there is enough cystine in the cell to synthesize glutathione (GSH). GSH is an essential substrate for GPX4 to exert its antioxidant function. When lipid peroxidation occurs in cells, GPX4 uses the reducing ability of GSH to remove cytotoxic lipid peroxides and reduce the content of reactive oxygen species (ROS), thereby preventing membrane oxidation and maintaining the stability of the membrane lipid structure, thereby protecting the lipid membrane and inhibiting cell ferroptosis.44 Sun et al.45 demonstrated through in vitro experiments that dexamethasone can inhibit the activity of the GSH antioxidant system in bone cells by downregulating the expression of SLC7A11/GPX4 and increasing the content of ROS, thereby causing excessive ferroptosis of bone cells. Shao Xuekun et al.46 demonstrated that antler peptide inhibits dexamethasone-mediated osteoblast ferroptosis by activating the SLC7A11/GPX4 pathway, providing a novel and promising therapeutic option for the treatment of SONFH.
6. Oxidative Stress Signaling
Steroid-induced osteonecrosis of the femoral head (SONFH) happens when there are too many reactive oxygen species (ROS) that damage cells and cause osteoblast and endothelial injury. The ROS–JNK–c-Jun signaling pathway is very important for controlling the autophagy and death of osteoblasts caused by glucocorticoids. When ROS levels rise, they activate JNK, which phosphorylates c-Jun, causing cells to die. However, crocin and lithium have been proven to stop this process and protect bone cells.8,26 Chen et al. found that glucocorticoids increase the levels of NADPH oxidase isoforms NOX1, NOX2, and NOX4 in human SONFH samples. For NOX1, the levels went from 37.6 ± 9.2% to 64.5 ± 7.6%, for NOX2, from 49.6 ± 16.5% to 90.8 ± 2.9%, and for NOX4, from 40.7 ± 6.8% to 85.5 ± 9.3% compared to controls with developmental dysplasia (Tan et al., 2021). Zhao et al. showed that NOX2 expression was 1.5 times higher in rat models of methylprednisolone-induced ONFH. They also found that 8-OHdG and TUNEL indices were two to three times higher, and empty-lacunae rates went from 9.4 ± 2.0% to 25.9 ± 4.7% (Zhao et al., 2015). The Keap1–Nrf2 antioxidant axis is highly crucial for keeping cells from getting too much ROS from glucocorticoids. When compounds like taxifolin and Letifinin turn on Nrf2, they make antioxidant enzymes like- HO-1 and NQO1 work harder and raise SOD production. This lowers osteoblast ROS and apoptosis and brings trabecular BV/TV back to normal in SONFH models.24,45 Chen et al. showed that selenium-doped silica nanocomposites (Se@SiO₂) release selenium in a regulated way, which raises SOD and GSH levels and greatly lowers ROS-mediated damage to the femoral head without affecting the therapeutic effect of methylprednisolone (Chen et al., 2020). N-acetylcysteine (NAC) is a well-known antioxidant and ROS scavenger that has been found to lower oxidative stress caused by glucocorticoids and keep osteoblasts from dying in preclinical models of SONFH. Luo et al. showed that NAC treatment lowered ROS levels inside cells, stopped JNK/c-Jun phosphorylation, and kept osteoblasts alive in cells that had been exposed to dexamethasone. This suggests that NAC could be used as a treatment for steroid-induced bone injury.8
7. Necroptosis
Necroptosis of bone microvascular endothelial cells (BMECs) makes microvascular impairment in SONFH worse. Bertheloot et al. explained how glucocorticoids or TNF-α start necroptosis by autophosphorylating RIPK1 at Ser166, bringing in RIPK3, and phosphorylating MLKL at Ser345. This causes p-MLKL to oligomerize, generate membrane pores, and kill endothelial cells.27
Velchov et al. showed that necrostatin-1 treatment in rat SONFH models cut the number of cases of the illness from 70% to 30%. It also kept BV/TV and trabecular thickness the same and cut p-RIPK1 and p-RIPK3 levels by about 50%.6 Yan et al. found that luteolin lowers the levels of p-RIPK1/RIPK1, p-RIPK3/RIPK3, and p-MLKL/MLKL ratios that dexamethasone causes to rise by 40% to 60% in BMECs. This brings back proliferation and tube formation in vitro.35
Because oxidative stress and necroptosis work together, ROS activates RIPK1. Combination therapies that combine Nrf2 activators like taxifolin with necroptosis inhibitors like necrostatin-1 or luteolin provide more protection than monotherapies in SONFH models.35,45 These therapies reduce both apoptotic and necroptotic cell death and better protect bone microarchitecture. Future research should look into the long-term safety, best dose, and therapeutic application of these techniques that combine antioxidants and necroptosis blockers.
8. Pyroptosis
Pyroptosis is a pro-inflammatory programmed cell death that forms pores in the cell membrane, causes continuous cell swelling, and eventually ruptures the cell membrane, releasing a large amount of inflammatory substances.47 On the one hand, moderate pyroptosis can induce inflammatory and immune responses, eliminate pathogenic microorganisms in the body, and prevent infection. On the other hand, excessive pyroptosis can not only inhibit the body’s immune defense function but also lead to bone metabolism disorders and induce aggravation of SONFH.48 Current studies have found that pyroptosis pathways mainly include the classical pathway mediated by Caspase-1 and the non-classical signaling pathway mediated by Caspase-4/5 (humans) or Caspase-11 (mice).
The NLRP3/Caspase-1 signaling pathway is a classic inflammasome pathway for cell pyroptosis. NLRP3 is an important intracellular pattern recognition receptor. Under the stimulation of various damage-related molecular patterns, it binds to the Caspase-1 precursor through the adaptor protein to form the NLRP3 inflammasome, which is then processed into activated Caspase-1. The latter can not only activate inflammatory factors such as IL-1β and IL-18, but also further activate the immune inflammatory response, causing cell damage. It can also cut and separate the N-terminal structure (NT) and C-terminal structure (CT) of the pore-perforating protein GSDMD, so that the activated NT without CT inhibition inserts into the cell membrane to form a membrane pore, destroying the integrity of the cell membrane, thereby allowing mature IL-1β, IL-18 and cell contents to flow out of the cell, enhancing the body’s inflammatory response and promoting cell pyroptosis.49,50 The LPS/Caspase-4/5/11 signaling pathway is a non-classical signaling pathway for cell apoptosis. Under the influence of LPS, it directly acts on its downstream target molecules to promote cell pyroptosis. In preclinical investigations, MCC950, a selective NLRP3 inflammasome inhibitor, has been shown to be effective at lowering inflammasome-mediated pyroptosis in bone cells. This helps to reduce steroid-induced osteonecrosis.51 It can also amplify the NLRP3/Caspase-1 classical pathway to promote cell pyroptosis52 further. Fang et al.51 found that fibroblast growth factor 23 (FGF23) can inhibit the NLRP3/Caspase-1 signaling pathway, promote the osteogenic differentiation of bone marrow mesenchymal stem cells and the angiogenesis of human umbilical vein endothelial cells, thereby alleviating SONFH. However, whether FGF23 is expected to be an important target for the treatment of SONFH requires further research. Ferroptosis, oxidative stress, necroptosis, and pyroptosis are also thought to have a role in the development of SONFH, along with autophagy and apoptosis. Table 3 shows the most important parts of these pathways.
9. Summary and Future Directions
Steroid-induced osteonecrosis of the femoral head (SONFH) happens when glucocorticoids damage the endothelium, lipids are not broken down properly, blood clotting is not working right, and a complicated network of programmed cell death pathways, such as autophagy, apoptosis, ferroptosis, necroptosis, oxidative stress, and pyroptosis, all of which lead to impaired bone formation, blood vessel damage, and bone death. Autophagy has a dual nature that depends on the situation. For example, AMPK/mTOR, PINK1/Parkin, and SIRT1/FoxO1 cascades protect cells and allow them to survive low levels of GC exposure. On the other hand, PI3K/Akt/mTOR-driven suppression or chronic overactivation causes osteoblast and endothelial cell death unless agents like lithium or rapamycin finely tune the process.
The balance between apoptosis and survival of osteoblasts depends on Wnt/β-catenin signaling, which keeps osteoblasts alive, and STAT1/caspase-3 and OPG/RANKL/RANK axis, which cause osteoclastogenesis and osteoblast death. Ferroptosis causes lipid peroxides to build up by downregulating SLC7A11 and GPX4. Antler peptides and GPX4 stabilizers can stop this. Too much ROS from NOX isoforms turns on JNK–c-Jun autophagy death signaling. Nrf2 activators like taxifolin, NAC, and selenium nano-carriers bring redox equilibrium back to normal. RIPK1/RIPK3/MLKL necroptosis hurts bone microvasculature, whereas necrostatin-1 and luteolin protect microcirculation. NLRP3/Caspase-1 and non-canonical caspases cause pyroptosis, which makes inflammation worse. MCC950 and FGF23 antagonists stop inflammasome activation.
A detailed picture of how autophagy, apoptosis, ferroptosis, necroptosis, and pyroptosis interact with each other at clinically relevant GC doses can help us find “therapeutic windows” for pathway-specific control. Combination treatments that use Nrf2 activators with RIPK1 or NLRP3 inhibitors and autophagy modulators show promise for protecting bone cells and microvasculature in a way that works together. For real-time risk classification and treatment monitoring, circulating biomarkers such as autophagy-related microRNAs, lipid peroxidation products, and endothelium extracellular vesicles need to be tested. Early-phase clinical trials of potential drugs (MCC950, antler peptides, puerarin, necrostatin-1, and FGF23 inhibitors) in flexible, biomarker-guided designs will help speed up decisions about whether to move further or not. Finally, delivery platforms that use nanotechnology, such as bone-targeted selenium or taxifolin nano-carriers, should be improved so that they can release drugs in a controlled way over time and space in the femoral head microenvironment. This will make the drugs work better locally and cause less harm to the whole body. In general, these strategic directions will lead to individualized, multi-modal treatments that can stop or slow down SONFH before it causes permanent damage to the joints.
The review offers a modern mechanistic summary of SONFH pathophysiology while recognising many methodological constraints that diminish trust in its findings. It did not conduct a thorough risk-of-bias or quality assessment utilising conventional instruments, hence undermining the evaluation of internal validity. The majority of the included research were exploratory laboratory or mechanistic in character, including animal models or cell-based experiments, with only a limited number of observational studies. Furthermore, the absence of standardisation in preclinical bias evaluation compelled the authors to utilise a narrative synthesis instead of formal bias ratings. Language bias (exclusively including English) and publication bias (relying on published literature that may favour significant results) have the potential to alter the evidence base and its application. The studies exhibited heterogeneity in design and results, so obstructing meta-analysis and precluding quantitative evaluation of effects or consistency; funnel plots or alternative bias tests were impracticable. So, the conclusions are a qualitative combination of mechanical ideas instead of statistically supported effect estimates. The authors stress that SONFH pathogenesis is changing and that future systematic reviews with more similar sample sets should use strict quality and bias checks and, when possible, look at quantitative synthesis. Would you like this to be rewritten as a single, polished paragraph that you can use in your proposal, or would you like it to be modified to highlight methodologies and biases for certain sections?
Funding
This work was supported by the Gansu Provincial Youth Science and Technology Fund (20JR10RA007).
Conflict of interest
The authors declare that they have no conflict of interest.
Authors’ contributions
All authors made substantial contributions to this work and have approved the final manuscript. NIU Xiaojuan and YAN Zhongsheng were responsible for the study conception and design. The initial draft of the manuscript was written by NIU Xiaojuan and CHEN Tiantian. LI Jun and YAN Xiangyong contributed to methodology, data acquisition, and analysis. WANG Wei assisted with software and visualization. WANG Jian provided supervision. All authors participated in reviewing the manuscript, critical revision for important intellectual content, and final approval.