1. Introduction
Osteoarthritis (OA) represents one of the leading causes of disability and functional impairment among working-age adults and the elderly population.1 According to the Global Burden of Disease Study 2019, the global prevalence of OA has dramatically increased by 113.25%, rising from 247.51 million in 1990 to 527.81 million in 2019.2 OA represents a huge challenge for healthcare systems, accounting for an estimated 1-2.5% of the gross national product in developed countries, and projected to quadruple by 2030.3,4 Within OA phenotypes, Hip Osteoarthritis (HOA) is the second most common subtype after knee OA (KOA), and is characterized by progressive cartilage loss, subchondral bone remodeling, and synovial inflammation.5 Although risk factors including biomechanical stress, advanced age, obesity, and altered joint loading are well-established, many individuals exposed to these factors do not develop OA.6,7 In the absence of secondary causes such as femoroacetabular impingement (FAI), post-traumatic arthritis, rheumatoid arthritis, or osteonecrosis of the femoral head, many HOA cases remain idiopathic. This strongly suggests that systemic mechanisms contribute to disease initiation and progression.8 Traditionally considered a “non-inflammatory” degenerative disorder, OA is now increasingly recognized as a condition characterized by chronic low-grade inflammation.9 Recent evidence suggested that this systemic inflammatory state may be influenced by the gut microbiome, defined as the collective genome and metabolic products of trillions of intestinal microorganisms.10,11 Dysbiosis, or disruption of the gut microbial balance, may compromise intestinal permeability, leading to systemic translocation of bacteria components such as lipopolysaccharides (LPS) and short-chain fatty acids (SCFAs). These molecules may stimulate immune responses, trigger pro-inflammatory cascades, and ultimately promote pathological bone remodeling.12–14 The role of gut-joint axis has been progressively acknowledged in OA, where altered microbial composition has been associated with increased synovial inflammation, pain sensitization, and accelerated joint degeneration.15 Given the pathophysiological features among OA phenotypes, similar microbiome-related mechanisms may contribute to idiopathic HOA, potentially influencing immune responses, cartilage homeostasis, and subchondral bone turnover, despite its deeper anatomical location. Increasing our knowledge of the gut microbiota and the role it may play in the pathogenesis of HOA could improve our understanding of disease mechanism, enable a more personalized risk assessment, and open the way to new therapeutic strategies and approaches for management and prevention of OA. While the “gut–joint axis” has been primarily characterized in knee osteoarthritis, this review extends the concept to the hip joint, introducing the notion of a “gut–hip axis” to describe the potential site-specific manifestation of systemic microbial influences. Due to the exploratory nature of the subject and the lack of hip-specific evidence, this manuscript was conducted as a narrative review and does not include a systematic search, PRISMA flow diagram, or formal risk-of-bias evaluation. To our knowledge, this is among the first review specifically focusing on the emerging link between dysbiosis and HOA, aiming to explore current evidence and outline future research directions in this promising and evolving field.
2. Methods
This narrative review was conducted to summarize the current evidence regarding relationship between the gut microbiota dysbiosis and HOA. A literature search was performed using PubMed, Scopus and Web of Science databases. The following search terms and combinations were used: “hip osteoarthritis”, “osteoarthritis”, “gut microbiome”, “gut microbiota”, “dysbiosis”, “gut–joint axis”, “gut–hip axis”, “short-chain fatty acids”, “inflammation”, and “microbiota-targeted therapy”. Relevant preclinical and clinical studies, systematic reviews, and randomized controlled trials were considered. Given the limited HOA- specific evidence available, studies focusing on KOA models and broader gut-joint axis were also included to support the biological rational of this review. Only articles published in English and considered relevant to the scope of the review were included.
3. Pathophysiology of Hip Osteoarthritis
The pathogenesis of early HOA is complex, multifactorial, and not yet fully elucidated. Physiological loading is essential for maintaining joint tissue homeostasis,16,17 but excessive or abnormal mechanical stress disrupts the balance between synthesis and degradation, leading to progressive cartilage degeneration.18–21 Repetitive shear stress induces a reduction in type II collagen and proteoglycan synthesis, an increase in pro-inflammatory mediators, and chondrocyte apoptosis.21 Within the osteochondral unit, subchondral bone remodeling with increased porosity and microcracks enables biochemical and mechanical communication between cartilage and bone.22–24 In this bidirectional exchange, cartilage releases cytokines and osteoclastogenic factors,22,25 while subchondral osteoblasts contribute to cartilage degradation.26 Early synovitis27 further supports the modern concept of OA as a whole-joint disease from its earliest stages. From an etiological perspective, HOA arises from both local, joint-specific factors and systemic influences that modulate the biological response to mechanical stress. Local morphological abnormalities alter joint load distribution and predispose to tissue degeneration. In developmental dysplasia of the hip (DDH), a shallow acetabulum increases load on the acetabular rim, and cartilage injury.20 In FAI, cam deformities induce anterosuperior delamination, while pincer variants cause labral damage due to acetabular over coverage.28,29 These morphological changes exist along a continuum, with severe deformities linked to early-onset OA and milder variants associated with primary late-onset OA.30 Periarticular muscle weakness and labral tears further exacerbate abnormal joint loading, accelerating cartilage degeneration.31–34 Systemic factors also influence joint susceptibility. Aging reduces tissue repair capacity,35–38 while hormonal changes may explain sex-specific trend: HOA is more frequent in men under 50 years but rises in postmenopausal women, with estrogen therapy may exert protective effects.18,39 While occupational exposure to heavy manual labor, particularly farming, increases the risk of HOA in predisposed individuals, no consistent association has been found with sports participation.40,41 Twin studies have shown that genetic factors may account for up to 60% of HOA risk,42 and ethnic differences suggest that morphological and genetic variations may be protective in some populations.43 Obesity represents a dual contributor: each 5-unit increase in BMI is associated with an approximately 11% increase in HOA incidence.44 Beyond mechanical overload, adiposity contributes to chronic low-grade inflammation, through adipokines and cytokines, affecting cartilage and bone metabolism.45 The link between obesity and OA in non–weight-bearing joint, such as the hands, further supports a systemic inflammation mechanism.46
4. The Gut–Joint Axis and KOA: A Paradigm for Understanding HOA
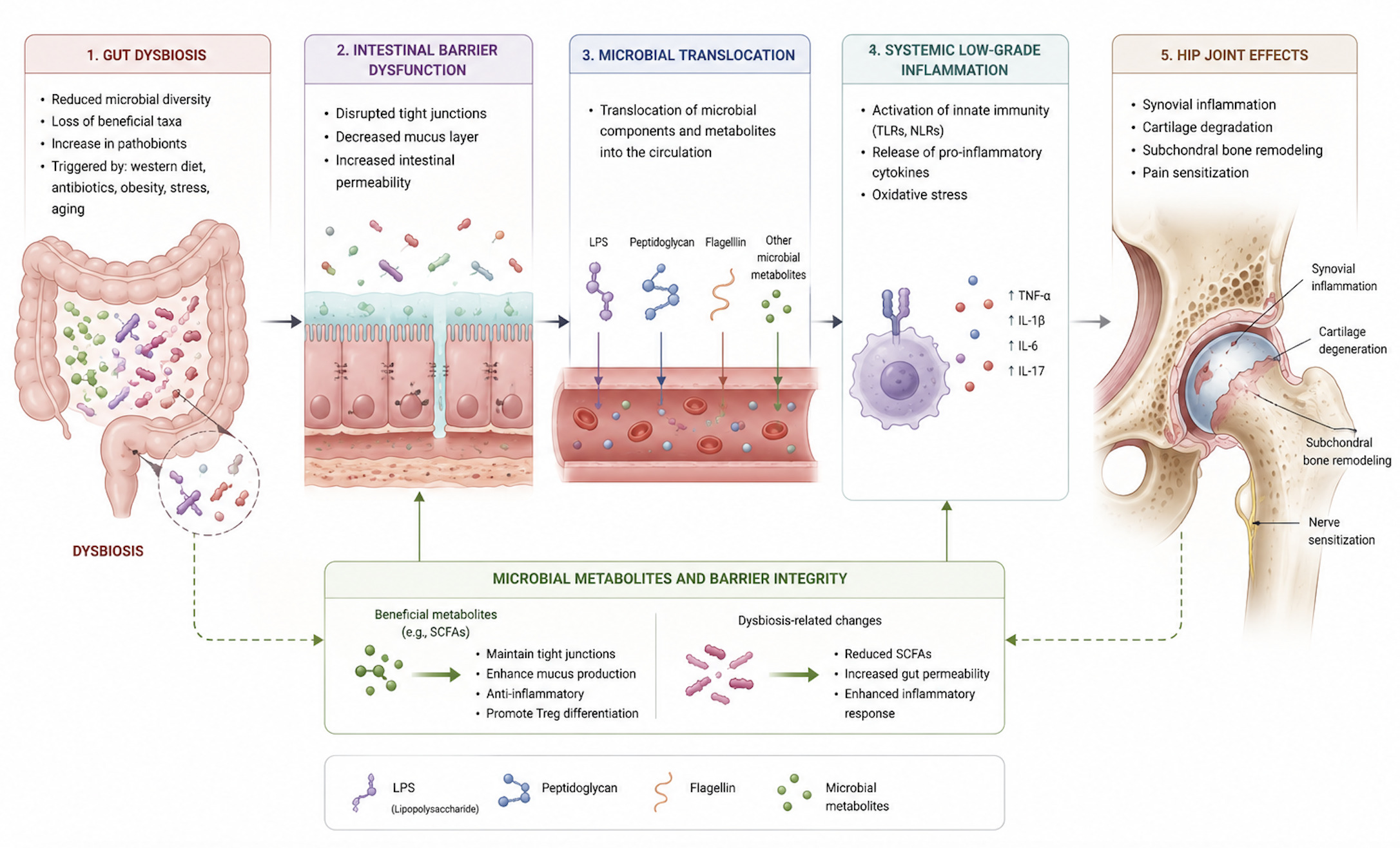
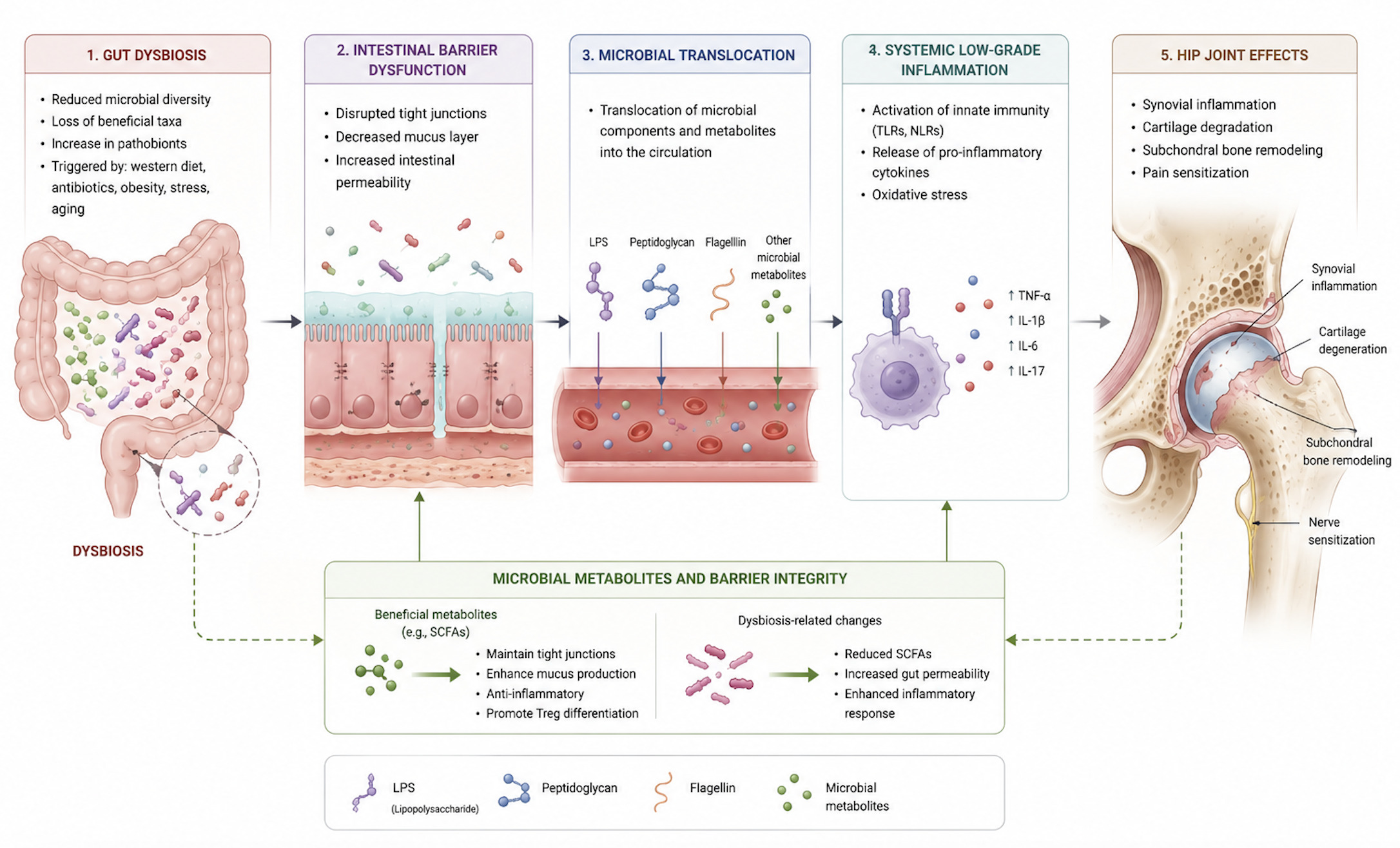
Idiopathic OA is no longer regarded as a purely mechanical disorder but as a disease of chronic low-grade inflammation. Even before overt structural damage, chondrocytes exposed to chronic inflammatory and metabolic stress shift toward glycolysis, increasing reactive oxygen species, suppressing autophagy, and upregulating matrix-degrading enzymes.47 Synovial macrophages in OA show an imbalance toward the pro-inflammatory M1 phenotype, driving tissue degradation and joint pain. Cytokines such as IL-6 and chemokines like CCL2, along with transcriptional regulators including NF-κB and LEF1, coordinate the catabolic and neurogenic responses that characterize the OA microenvironment.48 RNA-binding proteins (RBPs) have emerged as post-transcriptional regulators of inflammation, matrix remodeling, and chondrocyte senescence.49 Extracellular vesicles (EVs) further amplify degeneration by transferring cytokines, MMPs, and microRNAs between chondrocytes, synoviocytes, and joint-resident cells promoting apoptosis, and synovial activation.50 Over the last decade, a growing body of literature has emphasized the importance of the gut-joint axis, highlighting the potential impact of the intestinal microbiome on musculoskeletal tissues. Evidence from both preclinical and clinical studies has outlined a cascade linking intestinal dysbiosis, impaired barrier permeability, systemic dissemination of microbial products, immune activation, synovial inflammation, and pain sensitization. Most of the mechanistic and clinical evidence currently available derives from knee osteoarthritis (KOA) cohorts or animal KOA models. Therefore, the following concepts should be considered as extrapolations rather than confirmed hip-specific findings. Although both HOA and KOA share similar inflammatory pathways, anatomical and structural differences may influence the impact of microbial-derived inflammation. The hip joint demonstrates increased subchondral bone thickness and elevated microstructural porosity relative to the knee, which may promote microbial product transport and amplify subchondral immune activity.22,23 Moreover, the deeper vascular supply of the femoral head may increase exposure to circulating microbial metabolites or endotoxins in conditions of systemic dysbiosis. On the other hand, the knee joint may exhibit a quicker and more dynamic inflammatory response due to its higher synovial volume and more superficial vascular.25,51 These differences may explain subtle variations in how microbial-driven inflammation contributes to cartilage degeneration in HOA versus KOA.
4.1. Evidence from Animal Models of Dysbiosis
Animal models have provided some of the clearest mechanistic evidence linking dysbiosis to joint degeneration. Exposure to high fat/high sucrose (HFS) diets induces profound alterations in gut microbiota, characterized by loss of commensal Lactobacillus spp. and enrichment of pro-inflammatory Methanobrevibacter spp. Sun et al expanded these observations by showing that high-fat diet (HFD) compromises epithelial integrity, reduces tight junction proteins expression, such as zonula occludes 1 (ZO-1), and facilitates the translocation of LPS. In addition, bacterial outer-membrane vesicles (OMVs) may bypass mucosal defenses, delivering pro-inflammatory signals into the submucosa.52 Notably, visceral adiposity rather than overall body weight represents the strongest predictor of cartilage damage, highlighting the contribution of adipose-derived inflammatory mediators, including leptin, GRO-KC, MIP-1, IP-10, MIP-2, in both serum and synovial fluid. Increased intra-articular IL-1 levels further correlate with structural cartilage damage.53,54 These findings support the concept that dysbiosis is not merely an association but an active driver of joint pathology. Although animal studies are useful to explore mechanisms, most models induce dysbiosis with extreme diets, use very small sample sizes, and do not replicate typical human HOA exposures. These factors may limit internal validity and raise questions about external translation to human hip OA
4.2. Microbial Metabolites and Barrier Integrity
A central mechanism involves microbial metabolites, particularly short chain fatty acids (SCFAs), produced through microbial fermentation of nondigestible dietary fibers. Propionate and butyrate, generated by specialized taxa including Akkermansia muciniphila, Eubacterium dolichum, Ruminococcus bromii, Bacteroides spp, exerted protective effects on bone and joint tissues.55–58 SCFAs inhibit osteoclast differentiation by reducing pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6, as well as downregulating RANKL expression by B cells.59,60 Additionally, butyrate enhances osteoblast differentiation via T regulatory cells and stimulation of Wnt10b signaling.61,62 Furthermore, SCFAs contribute to maintain epithelial barrier integrity by reducing systemic inflammation and microbial translocation, thereby indirectly protecting against bone loss in inflammatory and postmenopausal osteoporosis.60,63,64 Ovariectomized mice analyses confirmed the potential of microbiota-targeted therapies by showing that Lactobacillus strains supplementation increases Claudin family protein expression, improves intestinal permeability, promotes osteoblast activity, and prevents sex steroid–induced bone loss.65,66 Preclinical and clinical researches have supported the important role that the microbiome plays in regulating skeletal homeostasis and contributing to bone-related diseases.60,67 He et al.68 demonstrated that decreased SCFA-producing taxa in postmenopausal women are associated with lower bone mineral density and higher bone resorption. In parallel, butyrate might modulate zonulin, a physiologic regulator of gut permeability. Increased permeability occurred in celiac disease or after bacterial stimuli. Guido et al. showed that reduction in the Bacteroidetes phylum impairs gut permeability. In this context, an abundance of Streptococcus spp increased LPS exposure, activates macrophages via NF-kB and the MAPK pathways, increases the M1/M2 macrophages ratio, and promotes production of MMPs and pro-inflammatory mediators. Chemokines such as the CCL2/CCR2 axis seem to be involved in the development of pain, while inflammation-induced upregulation of aquaporins (AQP1, AQP3) may trigger joint swelling in early OA, proteoglycans loss, and chondrocyte apoptosis.69 Such evidence supports the role of the gut microbiome in preserving bone health and modulating disease-related responses.
4.3. Immune Mechanisms and Inflammatory Response
An impaired gut barrier allows the translocation of microbe-associated molecular patterns (MAMPs), including LPS, peptidoglycan (PGN), flagellin, and bacterial DNA, which are able to activate innate immunity in distant tissues. By binding to pattern recognition receptors, especially Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), these molecules stimulate pro-inflammatory cascades in cartilage, bone, and synovium. Moreover, PGN may trigger NLRP3 inflammasome activation, leading to macrophages release of IL-1, and impaired cartilage extracellular matrix synthesis. Similarly, LPS engage the LPS–LBP–CD14–TLR4–MD-2 complexes, promotes NF-kB activation and upregulates TNF, IL-1, IL-6, IL-8, RANKL, and MMPs, ultimately accelerating cartilage degradation.
In addition, LPS may induce inflammation in adipose tissue, eventually stimulating the chronic inflammation state inside the knee joint.70–75 Gut permeability may also lead to transient bacteremia, whereby microbes or microbial products reach the joint either directly or via immune cells acting as “Trojan Horse”, as suggested by a recent systematic review.53
4.4. Gut Microbiome and Osteoarthritis
The presence of microbial DNA within joint tissues remains among the most debated findings. Dunn et al. identified distinct microbial communities in cartilage, with KOA samples enriched in Firmicutes and HOA characterized by the Proteobacteria.76 Similarly, other studies reported increased Clostridiales and decreased Lactobacillus spp. in OA joints.53 Although contamination cannot be excluded, the consistency of findings suggests the hypothesis of a resident joint microbiome. This concept remains highly debated. Because joint and cartilage samples are extremely low-biomass tissues, the detection of microbial DNA is inherently vulnerable to contamination from reagents, laboratory workflow, and environmental sources. Interestingly, Bonanzinga et al. reported that Cutibacterium, Staphylococcus, and Paracoccus spp. seem to be dominant in non-OA samples, whereas OA joints show higher Proteobacteria abundance.77 The divergence between hip and knee findings is particularly relevant, suggesting joint-specific microbial ecosystems that may differentially modulate disease pathogenesis.
The gut–joint axis is increasingly recognized as a potential driver of the pathogenesis of OA. As detailed above, SCFAs and other microbial metabolites may mediate protective effects by modulating inflammation and barrier integrity.78 Some microbial antigens may reach deep cartilage layers, triggering innate immune responses.79,80 Other microbial products may also influence chondrocyte epigenetics, altering DNA methylation and non-coding RNA profiles, especially in obesity-related OA.81 The microbiota influences systemic aging, oxidative stress, and circadian rhythms, all of which are implicated in joint degeneration82; moreover, circadian misalignment alters microbial composition, while dysbiosis reciprocally impairs circadian regulation, thereby disrupting cartilage homeostasis.83,84 Beyond structural damage, dysbiosis also appears to modulate nociception. Data from the Rotterdam Study showed that the abundance of Streptococcus spp. correlates with increased WOMAC pain scores and MRI-detected effusion in patients with KOA.10 Recent systematic review supported the clinical association between pain severity and microbiome alterations in KOA.77 Furthermore, systemic inflammatory mediators, such as IL-6, MDC, and IP-10, are significantly associated with pain in both KOA and HOA.51
Although these findings have yet to be replicated in HOA, they raise the possibility of shared microbial influences across joint sites.76 Dietary findings reinforce these mechanisms.54 Similarly, Schott et al. found that in murine model of OA induced by medial meniscal destabilization, a high-fat diet leads to gut dysbiosis, increased intestinal permeability, and elevated systemic IL-1β and TNF-α levels, which accelerate cartilage degeneration.85 Treatment with the prebiotic oligofructose supplementation restores a healthier microbiota, reduces circulating LPS, and attenuates joint damage, without significant weight loss, suggesting microbiota modulation as a potential chondroprotective strategy independent of mechanical stress.
4.5. Gut Microbiome and Hip Osteoarthritis
Evidence from genetics, animal models, and observational studies supported a causal role of the gut microbiota in the pathogenesis of HOA. Mendelian randomization analyses identified specific genera such as Gordonibacter and Eubacterium (brachy group) as risk enhancers, while Senegalimassilia, Slackia, and Streptococcus are linked to reduced risk.86 Animal studies provided further insights. In a canine model of spontaneous OA, Cintio et al. observed decreased fecal alpha diversity, increased Megamonas and decreased Paraprevotellaceae, Porphyromonadaceae, and Mogibacteria, findings consistent with human studies.87 These changes may either drive or result from the OA process. Genetic approaches further underscored the causal links between microbiota and HOA. Li et al., using a two-step bidirectional Mendelian randomization strategy, identified a protective effect of Actinobacteria (notably Bifidobacteria) on OA risk, partially mediated through basal metabolic rate (BMR).88 Elevated BMR was associated with increased OA risk, providing a novel mechanistic link between microbiota composition, systemic metabolism, and joint health. Conversely, Lee et al found no significant associations between gut microbiota and OA after multiple testing corrections, citing methodological limitations such as reliance on fecal data and joint-site heterogeneity.89 Although Mendelian randomization analyses have been interpreted as supportive of a causal relationship between gut dysbiosis and OA, these findings must be considered with caution. In microbiome Mendelian studies in particular, weak-instrument bias, and multiple testing across numerous taxa may lead to overestimation of causality. Therefore, Mendelian randomization supports biological plausibility rather than definitive causality for hip OA. Beyond systemic effects, the concept of a joint-specific microbiome has gained traction. Goswami et al. showed that microbial compositions derived from OA joints differ from environmental and skin flora, even after intra-articular injections or surgery.90 The bacterial profiles identified in their study were distinct from those commonly found in the skin, not attributable to procedural contamination, supporting the concept of a resident intra-articular microbiome.90 Similarly, Fernandez-Rodriguez et al. demonstrated joint-specific microbial profiles in human knees, suggesting potential extension to the hip.91 These host-microbiota interactions may influence both the pathogenesis of OA and susceptibility to periprosthetic joint infections (PJI). Dunn et al. also identified distinct microbial DNA in hip cartilage, including Pseudomonas, Acinetobacter, and Streptococcus, with joint-specific differences in microbial profiles and immune marker correlations.76 These findings suggest that the microbiota may modulate local inflammation and cartilage integrity directly within the hip joint. Microbiome-host interactions have also been observed in obesity-related HOA. Anderson et al. found that obese patients undergoing anterior hip approach surgery have higher prevalence of Enterobacterales in the skin microbiota overlying the surgical site compared to normal-weight individuals (21.4% vs. 2.8%).92 Such alterations may contribute to increased PJI risk in obese individuals, highlighting another microbiota-related pathway of surgical vulnerability. The therapeutic potential of the gut microbiota is emerging as a promising strategy in the management of OA. In a double-blind, randomized controlled trial, Karim et al. demonstrated that 12 weeks of high dose multistrain probiotics supplementation significantly improved pain, WOMAC scores, and postural balance, correlating with reduced plasma zonulin.93 Similarly, in a larger trial involving 537 patients, Lei et al. demonstrated that 6 months of Lactobacillus casei Shirota supplementation improved WOMAC and VAS scores and reduced systemic inflammation (CRP levels).94 The growing interest in this field has prompted authors such as Pedersini et al. to outline a trial protocol for advanced HOA and KOA (Kellgren–Lawrence ≥ 3), although clinical outcome data are not yet available.95 Human evidence directly linking dysbiosis to hip osteoarthritis is still limited. Only a few studies have specifically investigated HOA, and their findings should be interpreted as preliminary. The available data suggest possible correlations, but the strength of evidence is currently insufficient to establish causality. These ongoing investigations will clarify whether microbiota-based therapies can be translated in clinical opportunity. Figure 1.
5. Therapeutic Implications
Given these mechanistic links, microbiota-targeted strategies have been explored as potential therapeutic interventions. Synbiotics combinations, including oligofructose-inulin, Lactobacillus. rhamnosus, and Bifidobacterium lactis, have been shown to restore beneficial taxa, increase tight junction protein expression, and reduce pro-inflammatory cytokines.96 Prebiotics such as oligofructose may also exert prophylactic effects on OA development, by reducing gut permeability, increasing the expression of tight junction proteins, and modulating macrophage infiltration into synovium.52 Collectively, these approaches highlight the translational potential of manipulating the gut microbiota to modulate OA pathogenesis. Given the shared risk factors between KOA and HOA, including aging, obesity, metabolic syndrome, and low-grade systemic inflammation, it is plausible that similar microbiome-mediated pathways contribute to hip disease. To date, these mechanistic pathways have not been systematically linked to hip-specific outcomes. Thus, KOA serves as a blueprint for extending these concepts.
6. Future Directions
The gut-bone axis is emerging as a promising frontier in understanding the pathogenesis of HOA. However, most evidence comes from KOA or non-joint-specific microbiome studies, and true HOA-focused datasets are sparse. Therefore, mechanisms discussed in this review should be interpreted as biologically plausible but not fully demonstrated. Specific microbial taxa, metabolites, and functional pathways involved in cartilage degeneration and subchondral bone remodeling may be identified through high-resolution microbiome profiling and immunologic analyses. Randomized controlled trials are urgently required to evaluate microbiota-targeted interventions, such as well-characterized probiotics, dietary regimens promoting SCFA production, synbiotic, and, in selected cases, fecal microbiota transplantation.
7. Conclusion
Hip OA remains a debilitating condition with complex and incompletely understood pathophysiology. Traditional risk factors such as age, mechanical load, and structural abnormalities only partially explain its onset and progression. Although most of the recent advances in microbiome research have been achieved in KOA, the strong pathophysiological overlap between these two major forms of the disease suggests that dysbiosis may also play a central role in hip joint. Emerging findings support the concept that dysbiosis could represent a systemic, modifiable risk factor for OA, opening new avenues for prevention and treatment. Microbiome profiling could help identify specific subgroups of patients, particularly those without conventional risk factors, who may benefit from targeted interventions. In this context, microbiota-modulating strategies already explored in KOA, including probiotics, and SCFAs-enhancing diets, may eventually be evaluated in HOA as a potential adjunctive approach to slow disease progression. Overall, although current evidence remains preliminary, the close analogy with KOA strongly supports the existence of a gut-hip axis. The concept of a gut–hip axis remains promising but unproven. Confirming and expanding these findings in HOA may significantly advance our understanding of its pathogenesis and open the way for innovative therapeutic perspectives.
Author Contributions
Conceptualization, A.F, A.D.M., L.A.D.B.; methodology, A.F., A.D.M., and L.A.D.B, A.B.; software, A.B. and A.Z..; validation, B.Z, and L.A.D.B; investigation, A.F, A.D.M. and A.B..; resources, F.R.P, A.Z, and C.C.; writing— original draft preparation, A.F, A.D.M, A.B.; writing—review and editing, F.R.P. A.Z., and C.C,; visualization, B.Z.; supervision, L.A.D.B., E.A, R.P.;. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.